


Mucopolissacaridose (MPS) VI, ou síndrome de Maroteaux-Lamy, é um distúrbio devastador, progressivo e heterogêneo, com patologias graves que afetam múltiplos órgãos. É causada pela atividade deficiente de arilssulfatase B (ASB), a enzima que cataboliza os glicosaminoglicanos (GAGs) sulfato de dermatano e sulfato de condroitina.1
Embora haja uma grande variabilidade na apresentação fenotípica da MPS VI,1,2 geralmente os pacientes são caracterizados como tendo doença de progressão rápida ou lenta:
Independentemente da taxa de progressão, a MPS VI não tratada pode evoluir ao longo dos anos e acarretar o seguinte3:
A MPS VI é uma condição clinicamente heterogênea que pode apresentar-se como uma doença acentuada no primeiro ano de vida ou como uma doença que evolui mais lentamente, com o aparecimento dos sintomas no decorrer de um período de tempo mais longo. No entanto, é importante reconhecer que a MPS VI manifesta sintomas de forma contínua, portanto não há parâmetros fixos para essas características de progressão da doença.1,2
MPS VI de progressão rápida, caracterizada no estudo de pesquisa transversal por Swiedler et al como resultados de níveis urinários da proteína GAG (uGAG) acima de 200 μg/mg,3 e que pode se manifestar no primeiro ano de vida com sintomas inespecíficos. Entre os 2 e 3 anos, os traços característicos se desenvolvem, incluindo o seguinte1,2,5:
MPS VI de progressão lenta, caracterizada como níveis de uGAG inferiores ou iguais a 200 μg/mg, pode não estar presente explicitamente, retardando assim o diagnóstico. Contudo, os pacientes desenvolvem morbidade significativa que pode ser limitante e de ameaça à vida. Seja de progressão rápida ou lenta, a MPS VI geralmente não causa déficits neurocognitivos, embora limitações físicas possam afetar o aprendizado e o desenvolvimento dos pacientes.3
A tabela abaixo descreve as complicações da MPS VI por sistema orgânico. A manifestação clínica associada à MPS VI é heterogênea, no entanto, todos os pacientes apresentarão progressão da doença.

Pacientes com MPS VI expressam taxas de crescimento acentuadamente mais baixas que seus correspondentes etários não afetados. O desvio no crescimento apresentado por pacientes com MPS VI pode ser um sinal de problemas secundários e deve suscitar a vigilância clínica quanto a questões como desnutrição, anormalidades endócrinas ou privação psicossocial.

A MPS VI é uma condição clinicamente heterogênea com um espectro de fenótipos que apresentam manifestação e taxas de progressão variáveis.2 A figura abaixo demonstra a grande variabilidade entre um paciente com progressão rápida e outro com progressão lenta. O paciente com progressão rápida exibe muitos dos diferenciais fenotípicos da MPS VI.
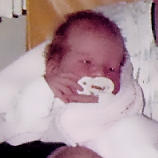
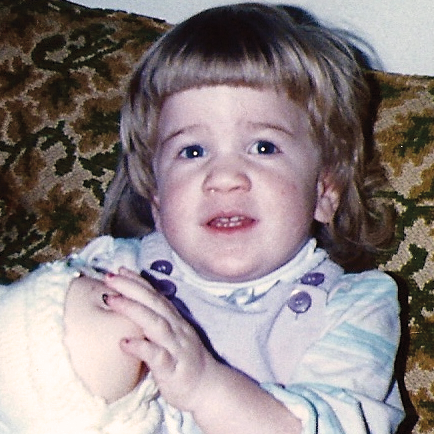


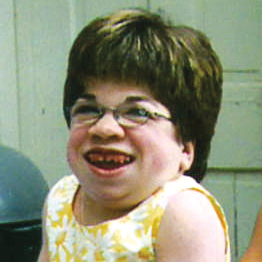
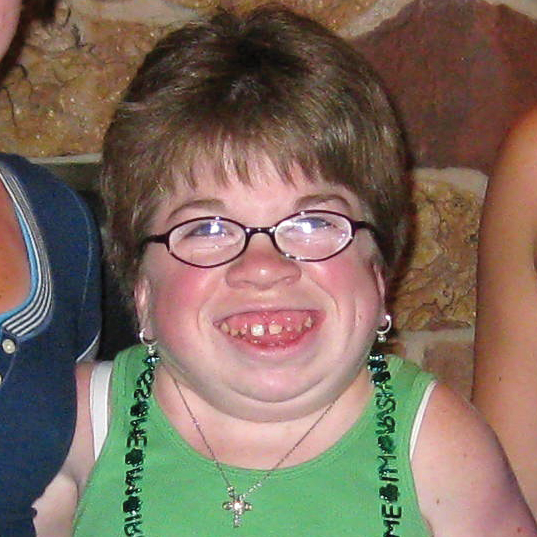
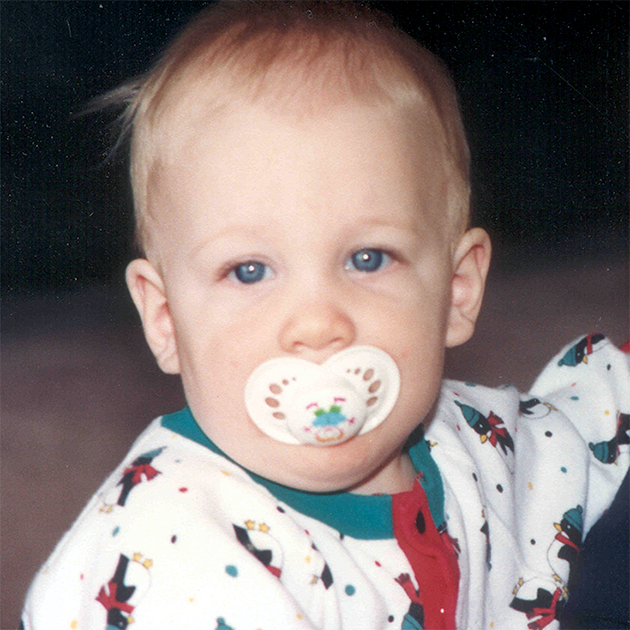

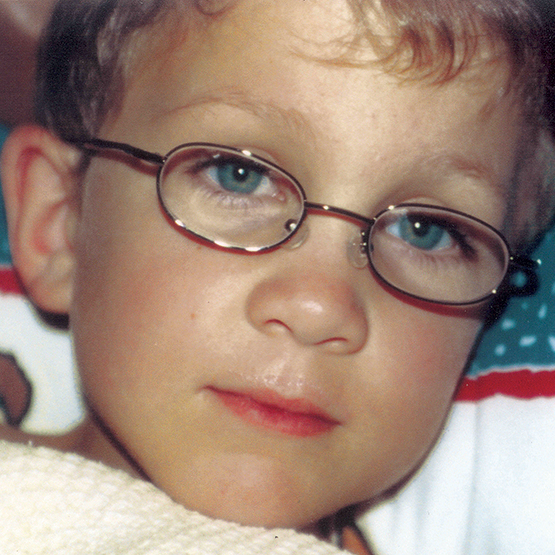



A distinção entre doença de progressão rápida e lenta somente é aparente em casos extremos. Na ausência de tratamento, a progressão da doença inibirá o bem-estar físico e funcional e resultará em uma expectativa de vida significativamente menor.4
Independentemente do fenótipo, os indivíduos afetados evoluem no decorrer de anos e, por fim apresentam o seguinte4
Pacientes com MPS VI geralmente morrem em decorrência de infecções, complicações secundárias à cirurgia ou doença cardiopulmonar.4
A MPS VI é uma doença autossômica recessiva causada por uma deficiência da enzima N-acetilgalactosamina 4-sulfatase (também conhecida como ASB). Essa deficiência resulta no acúmulo do GAG sulfato de dermatano nos lisossomos de uma ampla variedade de tecidos.13,14

Nenhum grupo étnico específico foi associado a um risco aumentado de MPS VI; no entanto, foram relatadas algumas frequências elevadas de mutações específicas. Há uma mutação comum (1533del23) encontrada em 23% dos alelos nos pacientes brasileiros com MPS VI. Adicionalmente, um estudo demonstrou uma alta prevalência de nascimento com MPS VI na população turca que vive na Alemanha, em comparação com a população alemão não turca.2
Ao longo da última década, o tratamento da MPS VI evoluiu juntamente com o conhecimento dos médicos sobre a doença, como por exemplo, sua variabilidade fenotípica e progressão. Outros fatores, como pacientes chegando à vida adulta, destacam novas áreas para pesquisa, entendimento e promessa no tratamento.
A MPS VI afeta múltiplos sistemas corporais, fazendo com que o tratamento de um espectro diverso das manifestações da doença seja uma parte importante da assistência integrada. O tratamento deve incluir o uso de dispositivos de adaptação ou de suporte, fisioterapia e terapia ocupacional, medicações com base nos sintomas, intervenções cirúrgicas e tratamento para repor a enzima deficiente.4
Publicadas em 2007, as “Diretrizes de Tratamento para Mucopolissacaridose” fornecem recomendações entre as especialidades para o tratamento de pacientes com MPS VI. Recomenda-se terapia de reposição enzimática (TRE) como uma opção de tratamento a ser considerada em pacientes com MPS VI.4
Além do início da TRE quando apropriado, as diretrizes recomendam estratégias de tratamento específicas do sistema para os sintomas associados ao acúmulo de GAG.4 O tratamento multiprofissional e multissitêmico e o tratamento com procedimentos oferecidos por uma equipe multidisciplinar de cuidados coordenados são componentes essenciais para otimizar os resultados dos pacientes.16 Os pacientes com MPS VI requerem avaliações precoces e regulares para analisar a progressão da doença em múltiplos sistemas orgânicos e para detectar e tratar possíveis complicações da doença.4 A tabela abaixo mostra o cronograma de avaliações para pacientes com MPS VI recomendadas pelas diretrizes de 2007.

Pacientes com MPS VI frequentemente requerem intervenção cirúrgica para tratar das complicações multissistêmicas da doença. Esse tratamento cirúrgico é complicado pela natureza da doença.16
Pacientes com MPS VI sofrem de múltiplos fatores que podem aumentar drasticamente o risco cirúrgico e a necessidade de monitoramento, incluindo o seguinte16:
Esses fatores complicam os cuidados cirúrgicos e anestésicos, exigem pré-planejamento e necessitam de técnicas específicas para a doença a fim de aumentar os resultados ideais.17
Procedimentos perioperatórios especializados durante a anestesia, como intubação e extubação e o uso de uma checklist de neuromonitoramento intraoperatório são essenciais para intervenções cirúrgicas bem sucedidas. Uma equipe cirúrgica integrada, consistindo em especialistas em MPS VI é crucial para resultados positivos e duradouros.16,17
À medida que os pacientes com MPS VI chegam à vida adulta, sua relação com a equipe médica mudará. Para ajudar nessa transição, são necessários planos individuais para minimizar as interrupções de tratamento, estender o apoio além do escopo de tratamento pediátrico e apoio para os pais e garantir que os pacientes adultos tenham conhecimento acerca do tratamento de MPS VI.18
Esses planos de transição devem ser adaptados às necessidades específicas de cada paciente, de forma que aqueles que conseguem se cuidar sozinhos tenham as ferramentas necessárias e aqueles que são mais limitados terão o atendimento e cuidados apropriados para apoiá-los. Os planos devem incluir uma avaliação para determinar a capacidade do paciente em atingir os objetivos definidos, bem como conhecimento e habilidade para comunicar as informações sobre sua condição.18
References: 1. Thümler A, Miebach E, Lampe C, et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis. 2012;35(6):1071-1079. doi:10.1007/s10545-012-9474-1. 2. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi:10.1186/1750-1172-5-5. 3. Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). Am J Med Genet A. 2005;134A(2):144-150. doi:10.1002/ajmg.a.30579. 4. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405-418. doi:10.1542/peds.2006-2184. 5. Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12(2):133-138. doi:10.1016/j.prrv.2010.10.005. 6. Lin H-Y, Chen M-R, Lin C-C, et al. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr Pulmonol. 2010;45(12):1205-1212. doi:10.1002/ppul.21309. 7. Kampmann C, Lampe C, Whybra-Trumpler C, et al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(2):269-276. doi:10.1007/s10545-013-9649-4. 8. Willoughby CE, Ponzin D, Ferrari S, Lobo A, Landau K, Omidi Y. Anatomy and physiology of the human eye: effects of mucopolysaccharidoses disease on structure and function—a review. Clin Exper Ophthalmol. 2010;38(suppl 1):2-11. doi:10.1111/j.1442-9071.2010.02363.x. 9. Ganesh A, Bruwer Z, Al-Thihli K. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol. 2013;24(5):379-388. doi:10.1097/ICU.0b013e3283644ea1. 10. Kantaputra PN, Kayserili H, Güven Y, et al. Oral manifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis. 2014;37(2):263-268. doi:10.1007/s10545-013-9645-8. 11. Quartel A, Hendriksz CJ, Parini R, Graham S, Lin P, Harmatz P. Growth charts for individuals with mucopolysaccharidosis VI (Maroteaux-Lamy Syndrome). JIMD. 2015;18:1-11. doi:10.1007/8904_2014_333. 12. Data on file. BioMarin Pharmaceutical Inc. 13. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013. 14. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584. 15. Kakkis ED. Enzyme replacement therapy for the mucopolysaccharide storage disorders. Expert Opin Investig Drugs. 2002;11(5):675-685. 16. Walker R, Belani KG, Braunlin EA, et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211-219. doi:10.1007/s10545-012-9563-1. 17. Vitale MG, Skaggs DL, Pace GI, et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333-339. doi:10.1016/j.jspd.2014.05.003. 18. American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians, Transitions Clinical Report Authoring Group, Cooley WC, Sagerman PJ. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics. 2011;128(1):182-200. doi:10.1542/peds.2011-0969.
 United States
United States Japan
Japan Brazil
Brazil Chile
Chile Mexico
Mexico