


 United States
United States Japan
Japan Brazil
Brazil Chile
Chile Mexico
Mexico


Reconheça os sinais, identifique a doença
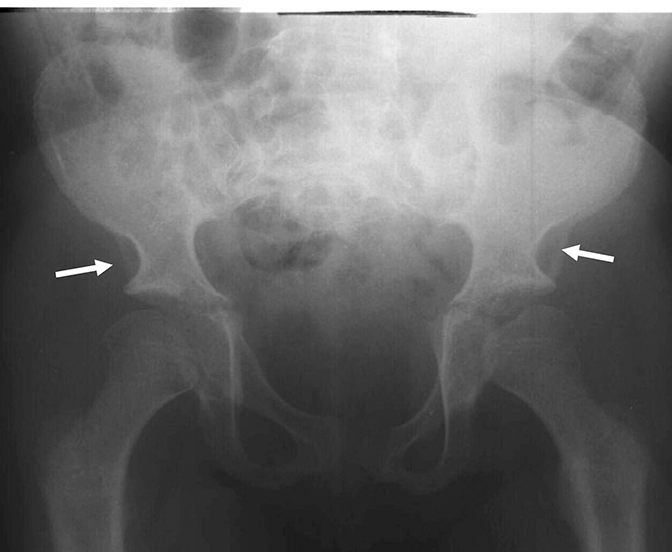
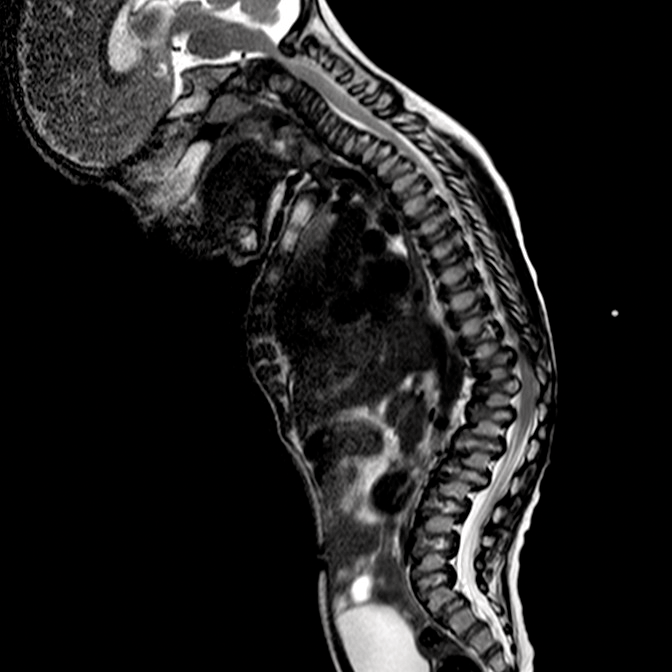
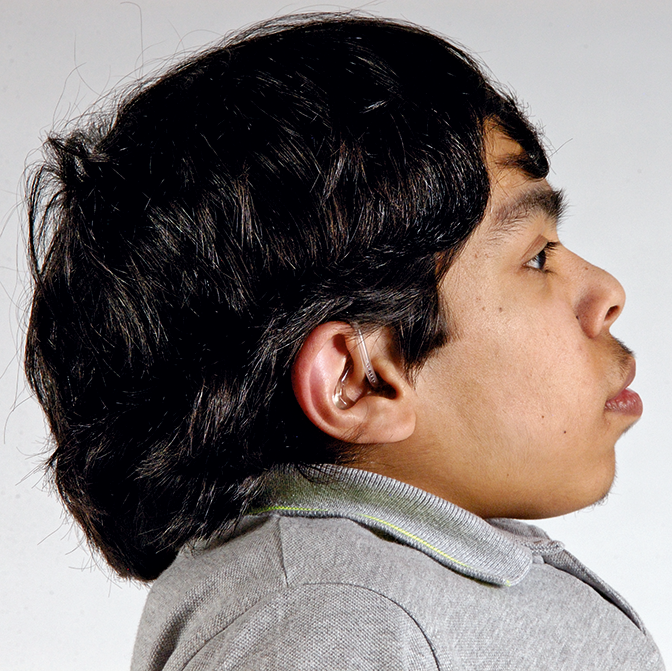
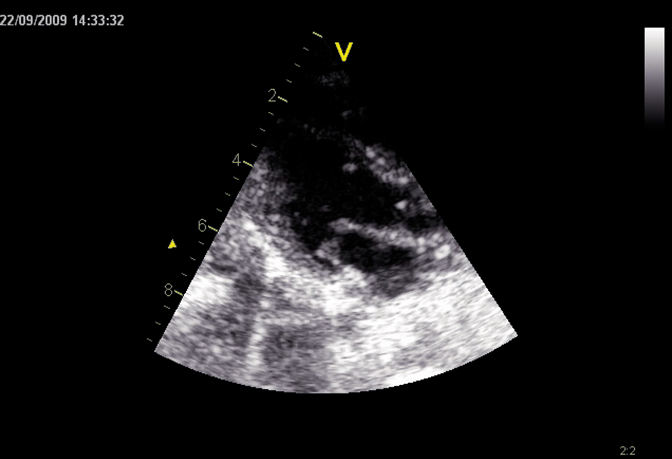
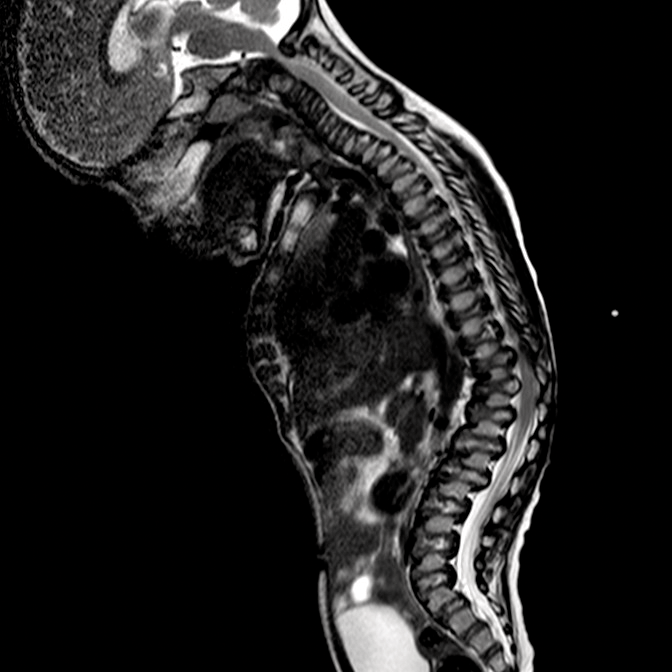
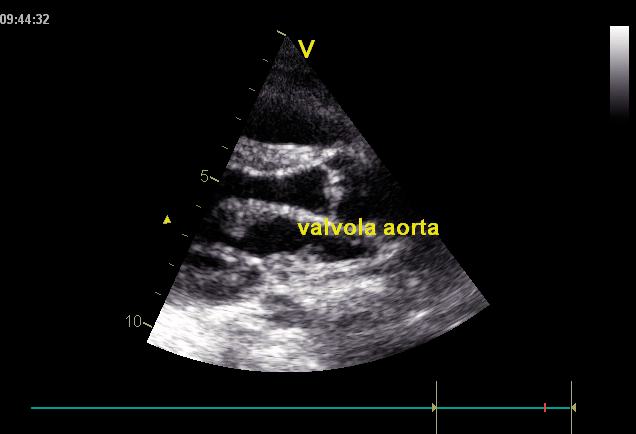
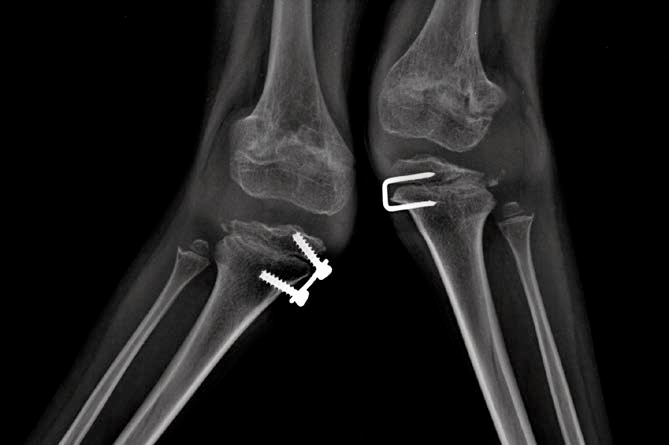
Os distúrbios da mucopolissacaridose (MPS) são um grupo de distúrbios sérios, herdados geneticamente e progressivos que afetam vários sistemas e funções corporais como resultado de deficiências enzimáticas expressadas de forma heterogênea.1-3 A variabilidade na apresentação e progressão da doença—assim como a raridade percebida da doença na prática clínica ampla-resultam no sub-reconhecimento da MPS nas práticas de especialidades médicas, levando a um diagnóstico tardio e a consequências potencialmente devastadoras.4,5 Otimize os resultados a longo prazo dos pacientes e ajude a iniciar o tratamento precoce reconhecendo os sinais da MPS e encaminhando os pacientes suspeitos a um geneticista ou centro metabólico imediatamente. O encaminhamento a um centro metabólico é a primeira etapa crítica no diagnóstico diferencial e início precoce do tratamento.4
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}