


Idade na ocasião do diagnóstico: 3 anos
Tempo até o diagnóstico: <1 anos
Principal sintoma diagnóstico: Murmúrio cardíaco
Médico que diagnosticou: Cardiologia pediátrica
Paciente com MPS VI, idades de 0 a 17
Descrição1:
Resumo
O diagnóstico rápido da MPS VI permitiu intervenção e tratamento precoces, os quais estão associados a melhores resultados clínicos.2
Os sintomas clássicos da MPS–baixa estatura, otite média recorrente, manifestações esqueléticas e reumatológicas–acompanhados de envolvimento cardíaco devem suscitar o encaminhamento a um geneticista ou centro especializado em metabolismo.2
Case history1
Terapia de reposição enzimática (TRE) iniciada





Descrição1:
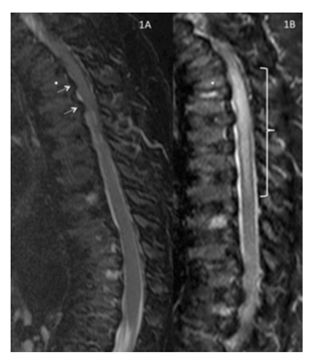
Recuperação de inversão sagital em T2/curta em T1–imagem ponderada das colunas torácica e lombar obtida 7 meses antes do procedimento.
Reproduzido com permissão de Drummond, Can J Anesth, 2015.
Resumo1
Neste caso ilustrativo, a lesão pós-operatória na medula espinhal foi desproporcional ao grau de estenose espinhal. Este caso é um exemplo da vulnerabilidade exagerada das pessoas com síndrome de Morquio A à compressão e ao dano da medula espinhal.
Os anestesiologistas devem considerar a estenose espinhal subclínica com frouxidão articular da coluna espinhal, além do risco de compressão.2
Histórico do caso1
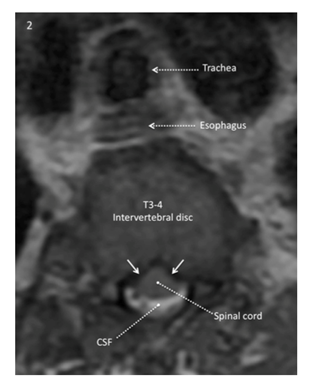
A imagem ponderada axial de T2 no nível do disco intervertebral T3-4 obtida 7 meses antes do procedimento mostra o líquido cefalorraquidiano (branco) ao redor da porção posterior da medula espinhal, mas não anterior. A superfície anterior da medula apresenta uma configuração convexa “rugosa” em vez de lisa, refletindo a pressão ventro-lateral bilateral sobre a superfície da medula (seta sólida branca).
Reproduzido com permissão de Drummond, Can J Anesth, 2015.
Baseado em um estudo de caso fornecido pelo Dr. Zakharchuk
Descrição1:
Abreviações: 3MSC, teste do degrau de 3 minutos; 12MWT, teste da caminhada de 12 minutos; FEV1, volume expiratório forçado em 1 segundo; FVC, capacidade vital forçada; GAG, glicosaminoglicano.
Resumo1
O diagnóstico precoce é essencial para iniciar a TRE, quando disponível, e proporcionar oportunidades de melhorar os resultados dos pacientes.3-6,b Como demonstra este caso, a TRE tem o potencial de melhorar parâmetros clínicos importantes, como a resistência e as medições respiratórias, o que pode ser essencial à qualidade de vida, a manutenção da capacidade de caminhar e as atividades rotineiras do paciente.7,8
Histórico do caso1
Idade no diagnóstico: 12 anos
Tempo até o diagnóstico: 11 anos
Principal sintoma diagnóstico: Atrasos motores e na fala
Médico que diagnosticou: Geneticista
Início dos sintomas neurológicos com 1 ano de idade, acompanhado por atraso no desenvolvimento de rápida progressão, levando ao diagnóstico de MPS IIIA em 2 irmãs1
Descrição1:
Resumo1
Conforme demonstrado neste caso, os sinais e sintomas podem progredir rapidamente, em particular com deterioração neurológica e agravamento do retardo comportamental em pacientes com MPS IIIA. Para as famílias com vários filhos demonstrando quaisquer características indicativas de MPS, o índice de suspeita deve ser especialmente alto.1
Embora o monitoramento e avaliação contínuos sejam cruciais, a intervenção precoce pelo encaminhamento direto a um geneticista e/ou centro metabólico, em vez de análise metabólica usando uGAGs, é o recomendado para um diagnóstico ágil.1
Histórico do caso1
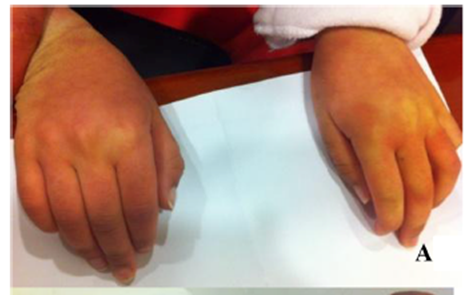
Examine revela descoloração cutânea evidente em paciente com MPS IIIA.1
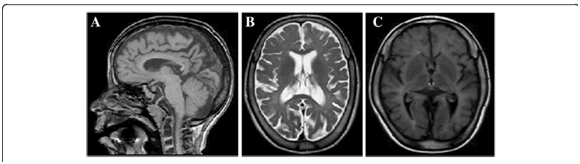
MRI revelou hipomielinação difusa, afinamento do corpo caloso e atrofia cerebral moderada, sintomas que se desenvolveram ao longo do tempo.1
References: 1. Data on file. Biomarin Pharmaceutical. 2. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 3. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)–10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584.
References: 1. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45-49. doi:10.1007/s12630-014-0247-1. 2. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983.
References: 1. Data on file. Biomarin Pharaceutical. 2. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 3. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi:10.1186/1750-1172-5-5. 4. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13-18. doi:10.1093/rheumatology/ker395. 5. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 6. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 7. Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology. 2011;50:v4-v12. doi:10.1093/rheumatology/ker394. 8. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 9. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63-72. doi:10.1016/j.ymgme.2013.11.015.
Reference: 1. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78.
 United States
United States Japan
Japan Brazil
Brazil Chile
Chile Mexico
Mexico